Accounting for Errors in Data Improves Divergence Time Estimates in Single-cell Cancer Evolution
Kylie Chen, PhD Candidate, School of Computer Science
Introduction
Single-cell sequencing provides a new way to explore the evolutionary history of cells. Compared to traditional bulk sequencing, where a population of heterogeneous cells is pooled to form a single observation, single-cell sequencing isolates and amplifies genetic material from individual cells, thereby preserving the information about the origin of the sequences. However, single-cell data are more error-prone than bulk sequencing data due to the limited genomic material available per cell. Here, we present error and mutation models for evolutionary inference of single-cell data within a mature and extensible Bayesian framework, BEAST2 [1].
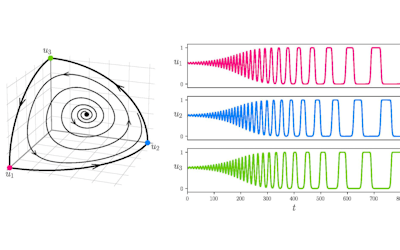
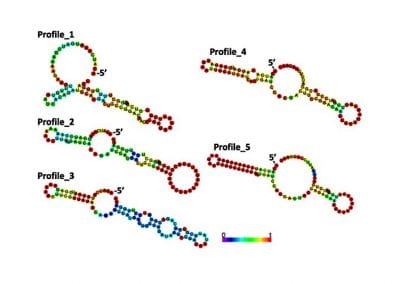
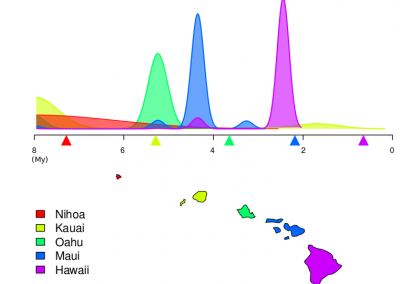
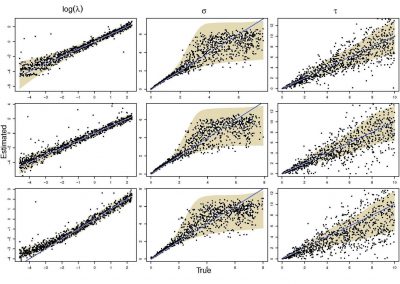
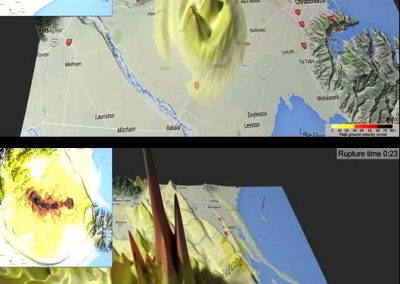
Our framework enables integration with biologically informative models such as relaxed molecular clocks and population dynamic models. Our simulations show that modeling errors increase the accuracy of relative divergence time and substitution parameters. We reconstruct the phylogenetic history of a colorectal cancer patient and a healthy patient from single-cell DNA sequencing data. We find that the estimated times of terminal splitting events are shifted forward in time compared to models which ignore errors. We observed that not accounting for errors can overestimate the phylogenetic diversity in single-cell DNA sequencing data as shown in Figure 1.
We estimate that 30–50% of the apparent diversity can be attributed to error. Our work enables a full Bayesian approach capable of accounting for errors in the data within the integrative Bayesian software framework BEAST2.
Use of Nectar GPUs
In our work, we provide a full Bayesian framework for inferring cell evolution histories and population dynamics using single-cell DNA data. We developed and optimized error models for single-cell DNA evolution. We present two error models for single-cell data: GT16 (allelic dropout/sequencing error) where parameters are sampled, and GT16 GL [2] where data uncertainty is based on phred scores from empirical single-cell variant callers. We performed Bayesian inference on single-cell DNA data using Monte Carlo Markov Chain sampling.
Our software was optimized to use a high performance GPU library, Beagle [3] for computing the likelihood probabilities. For analysis of large real datasets (25k sites, 26 cells), our GPU optimization can lead to a 30X decrease in computational time compared to the unoptimized code. The runtime efficiency for this dataset is 36 hours per million samples (no optimization), 4 hours per million samples (Beagle no GPU), and 1 hour per million samples (Beagle with GPU).
This work is published in Molecular Biology and Evolution, Volume 39, Issue 8, August 2022, msac143 (https://doi.org/10.1093/molbev/msac143)


Figure 1 Phylogenetic history of a colorectal cancer patient and a healthy patient from single-cell DNA sequencing data. p.s. The horizontal meaurement axis is applied to both top and bottom graphs.
References
- R. Bouckaert, J. Heled, D. Ku¨hnert, T. Vaughan, C.-H. Wu, D. Xie, M. A. Suchard, A. Rambaut, and A. J. Drummond, “Beast 2: a software platform for bayesian evolutionary analysis,” PLoS computational biology, vol. 10, no. 4, p. e1003537, 2014.
- A. Kozlov, J. M. Alves, A. Stamatakis, and D. Posada, “Cellphy: accu- rate and fast probabilistic inference of single-cell phylogenies from scdna-seq data,” Genome biology, vol. 23, no. 1, pp. 1–30, 2022.
- D. L. Ayres, A. Darling, D. J. Zwickl, P. Beerli, M. T. Holder, P. O. Lewis, J. P. Huelsenbeck, F. Ronquist, D. L. Swofford, M. P. Cummings et al., “Bea- gle: an application programming interface and high-performance computing library for statistical phylogenetics,” Systematic biology, vol. 61, no. 1, pp. 170–173, 2012.